Rapid advances in high-throughput genomic technology have enabled biology to enter the era of ‘Big Data’ (large datasets). The plant science community not only needs to build its own Big-Data-compatible parallel computing and data management infrastructures, but also to seek novel analytical paradigms to extract information from the overwhelming amounts of data. Machine learning offers promising computational and analytical solutions for the integrative analysis of large, heterogeneous and unstructured datasets on the Big-Data scale, and is gradually gaining popularity in biology. Our lab also working on developing machine learning models for data mining in plants, and the ultimate goal is to construct a Big-Data virtual environment for the plant research community. In a recent review in the journal TRENDS in PLANT SCIENCES, we systematically introduce some basic concepts and models for machine learning in plants.
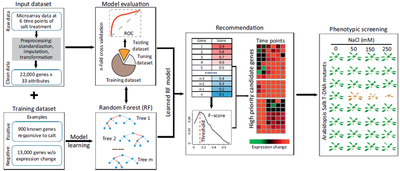
To explain the basic machine-learning terms and procedures for building a machine-learning system, we demonstrate the use of a supervised machine-learning method to discover genes responsive to salt stress and recommend high-priority candidate genes for phenotypic screening experiments (Figure I). The raw data are composed of 22 000 genes (examples) probed with an Affymetrix microarray whose expression values were profiled at six time points over a 24-h period, while root tissue was subjected to salt treatment. The raw data were first preprocessed by normalization to remove technical variations, imputation to replace missing values, and the transformation of six time-point values to 33 numeric statistics (attributes) to characterize the degree of expression change. Then, the clean dataset was converted to an input matrix of 22 000 genes in rows and 33 attributes in columns. The training set includes 900 known salt-related genes (labeled examples) as positive samples and 13 000 genes without expression change as negative samples. We treat this analysis as a classification problem, and use the random forest (RF) classifier (machine-learning model) to determine whether a gene was related to salt-induced response. The RF classifier builds a series of decision trees by resampling positive and negative samples, which are used to train and validate the final model. The n-fold cross-validation (evaluation metric) is used to assess the generation performance of the RF model (model validation and evaluation). The entire gene matrix is randomly split into n subsets; each of which keeps a roughly equal number of positive and negative samples. For each cross-validation, n–1 subsets are firstly merged into one dataset and then divided into two parts: the training dataset and the tuning dataset. Optimized parameters, at which the RF classifier has high precision and recall, are obtained by training on the training dataset and testing on the tuning dataset. The remaining subset is used as the testing dataset to evaluate the trained RF classifier’s prediction power by the ROC curve that plots the variation of TPR (y axis: true positive rate) versus FPR (x axis: false positive rate) at all possible prediction scores. After n rounds of cross-validation, an average AUC is calculated to represent the predictive power of the trained RF model. All the genes are ordered based on the priority scores assigned by the trained RF model. The genes with higher priority scores than a threshold are recommended as stress-related candidate genes, and the optimal threshold is determined when the maximal F-score is achieved. The recommended high-priority candidate genes with available Salk transfer DNA (T-DNA) mutation lines are functionally tested by phenotypic screening experiments.